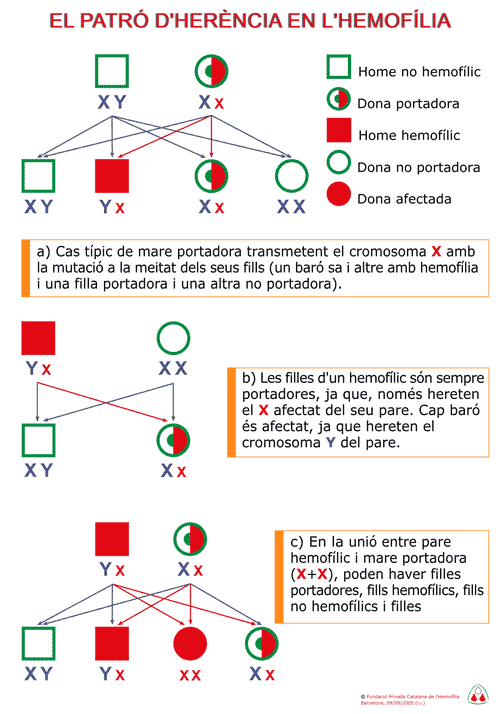
En aquesta figura s’il·lustra
la descendència familiar d’un
malalt hemofílic
i la d’una dona portadora. En el primer cas tots
els fills barons són normals i totes les filles
són portadores. En el segon cas la meitat de
les filles son portadores i la meitat dels fills presenten
la malaltia.
1.2. Tipus d’hemofília
Han estat descrits els dèficits congènits
de diversos factors de la coagulació (factor
II, factor V, factor VII, factor X, fibrinogen), però
identifiquem amb el nom d’hemofília A la
deficiència de factor VIII i amb el d’hemofília
B (també denominada malaltia
de Christmas) la
de factor IX. L’hemofília A és entre
4 i 5
vegades més freqüent que la B. Les
hemorràgies són més freqüents
i greus en l’hemofília A que en la B.
1.3. Gravetat de l’hemofília
S’han descrit tres nivells de gravetat
de l’hemofília (greu o
severa, moderada
i lleu) amb relació a l’activitat de factor
present en el plasma. Aquest fet és molt important
per dos motius:
è Generalment, però no sempre, el nivell
d’activitat de factor present en el plasma del
malalt determina la freqüència i
gravetat
de les manifestacions hemorràgiques, i en conseqüència defineix també les necessitats de tractament.
Així com menor sigui el nivell d’activitat
de factor en el plasma més fortes seran les hemorràgies
i augmentaran també els requeriments de factor
per tractar-les.
è El nivell d’activitat de factor i per
tant la gravetat de l’hemofília es manté
constant en una família determinada.
Així
el nadó hemofílic en el si d’una
família amb història d’hemofília
moderada tindrà també una forma moderada
de la malaltia.
Quan el nivell de factor és inferior al 1%,
parlem d’hemofília greu.
En aquests casos
les hemorràgies espontànies són
molt freqüents i greus. Sovint els malalts amb
hemofília greu presenten alteracions importants
de les articulacions com a conseqüència
de les repetides hemorràgies articulars.
Les petites quantitats de factor, entre l’1 i
el 5%, presents en els malalts amb hemofília
moderada són suficients per proporcionar una
certa protecció, de manera que les hemorràgies
espontànies són menys greus i menys freqüents.
Els malalts amb hemofília lleu tenen uns nivells
de factor
entre el 5 i el 25%. Aquests no presenten
hemorràgies espontànies i sovint se’ls
la diagnostica d’adults.
1.4. La coagulació
normal
L’hemostàsia és el conjunt
de mecanismes i reaccions que tenen com a finalitat
mantenir la sang en l’interior dels vasos sanguinis
impedint la seva sortida als teixits que l’envolten.
Aquest
fenomen té 4 fases: fase vascular, fase
plaquetària, fase plasmàtica i fibrinòlisi.
Fase vascular
Consisteix en un reflex nerviós que té
com a objectiu l’alliberació d’unes
substàncies que provoquen la contracció
del vas lesionat, amb la qual cosa disminueix el flux
de sang i en conseqüència l’hemorràgia.
Fase plaquetària
Es desenvolupa en dues etapes: l’adhesió
de les plaquetes al subendoteli vascular i l’agregació
de les plaquetes entre si. En aquesta fase és
molt important la participació del factor von
Willebrand, que estableix ponts d’unió
entre les plaquetes i els
vasos lesionats.
El factor von Willebrand és una proteïna
del plasma a la qual
està fixat el factor VIII.
La seva deficiència o absència dóna
lloc
a una altra malaltia hemorràgica coneguda
com a malaltia de
von Willebrand.
Altres proteïnes com la fibronectina i el fibrinogen,
així com el calci, són també necessaris
per al bon funcionament d’aquest mecanisme. Fase plasmàtica
És el conjunt de reaccions enzimàtiques
que tenen la finalitat
de convertir el fibrinogen, que
és una proteïna soluble, en fibrina, que és
insoluble. La fibrina té una estructura fibrilar
i
és la base del coàgul.
El conjunt d’enzims que intervenen en aquesta fase,
són els denominats factors de la coagulació,
que circulen en la sang
de forma inactiva i es converteixen
en formes actives per
l’acció d’un
altre factor prèviament activat.
En la figura s’observen les diverses reaccions
que tenen lloc
en aquesta fase, que es desencadenen
per dues vies:
- Intrínseca, quan la sang entra en contacte
amb el col.lagen subendotelial i un conjunt de proteïnes
que formen el que denominen sistema de contacte. La
cefalina és la prova de laboratori que analitza
aquesta via.
- Extrínseca, que s’inicia quan la sang
entra en contacte amb
el factor tisular, que és
una proteïna present en els teixits i
que és
alliberada quan hi ha una lesió vascular. La
prova de coagulació que explora la via extrínseca
és el temps de Quick
o taxa de protrombina. Fibrinòlisi
Al mateix temps que es desencadenen les reaccions
descrites,
es posen en funcionament els mecanismes que
tendeixen a dissoldre el coàgul amb la finalitat
que el vas lesionat torni a
ser permeable i la sang circuli
novament. Aquest mecanisme
es denomina fibrinòlisi
i es basa fonamentalment en l’activitat d’un
enzim, la plasmina, que degrada la fibrina.
Del perfecte equilibri entre aquests mecanismes depèn
que la sang es mantingui liquida a l’interior
dels vasos sanguinis, que únicament es formi
un coàgul quan hi ha una lesió vascular
i
que posteriorment els vasos tornin a ser permeables.
2. Manifestacions
clíniques i diagnòstic de l'hemofília:
Les manifestacions hemorràgiques del malalt
hemofílic estan
en relació amb la gravetat
de l’hemofília, i es presenten de forma
espontània en els hemofílics severs, i
solament després de traumatismes o en intervencions
quirúrgiques en els malalts moderats.
Les hemorràgies poden aparèixer a qualsevol
edat o en qualsevol part de l’organisme. La seva
gravetat dependrà del volum de l’hemorràgia,
de la seva localització i de la capacitat de
deixar seqüeles o lesions irreversibles.
2.1. Les hemorràgies
en el nadó
Fins a un 2% dels malalts poden presentar hemorràgia
intracranial durant el part. En alguna ocasió,
aquesta pot ser
la forma de presentació de la
malaltia, especialment quan no
hi ha altres antecedents.
Quan es fa un diagnòstic prenatal ha d’evitar-se
la vacuoextracció (extracció mitjançant
ventosa) del fetus ja que els hematomes cefàlics
poden produir anèmia i tarden a reabsorbir-se.
A vegades poden associar-se a hemorràgia cerebral.
Durant
els primers mesos, les manifestacions clíniques
són poc importants. Si cal practicar anàlisis,
les extraccions han de
fer-se amb la màxima cura
per evitar l’aparició d’hematomes
importants.
Els malalts han de rebre totes les vacunes pròpies
de l’edat i,
a més a més, les de
l’hepatitis A i B. Amb la dentició poden
aparèixer sagnats bucals però la complicació
oral més freqüent
és el sagnat per
ruptura del fre de la llengua durant la fase de desenvolupament
oral.
En començar a caminar, els hematomes de les
cames i dels
glutis són els més freqüents
i comencen a aparèixer les hemartrosis.
2.2. Localitzacions hemorràgiques
més freqüents
Hemorràgies mucoses:
Malgrat que generalment tenen poca gravetat,
generalment
també requereixen tractament. Les
més freqüents són les epistaxis (hemorràgia
nasal), les gingivorràgies (hemorràgia
per les genives), les secundàries a ferides per
mossegada de
la llengua o a la caiguda de la primera
dentició (dents de llet).
Especial gravetat poden tenir les secundàries
a ferides per
espines de peix en la faringe, ja que
poden produir un hematoma faringi amb obstrucció
de les vies aèries i en conseqüència
amb la impossibilitat de respirar normalment. Hematomes musculars:
Es produeixen espontàniament, per traumatismes
directes o
per esquinços en exercicis violents.
Es posen de manifest per dolor, augment del volum i de
calor local i impotència
funcional.
Les
lesions
musculars provocades per la presencia de la sang creen
una tendència a produir noves hemorràgies
del múscul danyat.
Atès que hi ha músculs encapsulats, la
tensió provocada per
la sang extravessada pot
produir la destrucció del múscul, la compressió
del sistema vascular (artèries i venes) i dels
nervis veïns amb conseqüències funcionals
importants. Això es coneix amb el nom de síndrome
compartimental, que es produeix fonamentalment en l’avantbraç
i en la cama.
Cal destacar l’hematoma del psoes, que es manifesta
per dolor en l’engonal i flexió del maluc,
i que sovint té tendència a reaparèixer. Hemorràgia cerebral:
Pot produir-se de forma espontània, si bé
generalment apareix després de traumatismes. Es
manifesta per la presència de mal
de cap, vòmits
i pèrdua del nivell de consciència. En ocasions,
amb deficiències motores i trastorns de la coordinació.
Requereixen tractament substitutiu immediat amb el factor
deficient i
en ocasions pot ser necessari també el tractament
quirúrgic.
Hemorràgia
articular:
Son les més freqüents en els malalts
hemofílics. Es caracteritzen
per dolor, augment
del tamany de l’articulació, calor i pèrdua
de moviment. L’articulació afectada queda
immobilitzada en la posició que permeti un major
volum de la cavitat articular.
La presència de sang en l’interior de
l’articulació, posa en
marxa una sèrie
de mecanismes que afecten la membrana sinovial i provoquen
la seva hipertròfia i la tendència a presentar
noves hemorràgies.
Els dipòsits de ferro i la inflamació
crònica de la sinovial
(sinovitis crònica
hipertròfica), afectarà també el
cartílag i l’os.
A la llarga
es produirà
una destrucció de l’articulació
amb pèrdua de mobilitat i, fins i tot, la seva
fixació en posicions vicioses. Per tal d’evitar
aquestes complicacions, és molt important el
tractament correcte de les hemorràgies articulars. Hemorràgia urinària:
Les hematuries espontànies no són
infreqüents en el malalt hemofílic. Generalment,
no cal un tractament específic, el
repòs
i una abundant hidratació són suficients.
En ocasions, quan apareixen de forma secundària
a la presència de litiasi (càlcul
o pedra),
cal un tractament urològic específic.
Hemorràgia digestiva:
Són produïdes per lesions digestives
diverses típicament hemorràgiques: hèrnia
de hiatus, ulcus, les varius esofàgiques, tumors
i malalties ulceratives, com la malaltia de Crohn i la
colitis ulcerosa.
Es posen de manifest per la presència de hematèmesi
(vòmit de sang), melenes (sang fosca en la femta)
i rectorràgies
(sang vermella en femta). A més
del tractament substitutiu, amb factor i si cal també
amb sang, és indispensable diagnosticar el focus
hemorràgic generalment mitjançant endoscòpia. Hemoptisi:
L’aparició d’hemoptisi no és
més freqüents en els malalts hemofílics,
però poden ser més greus. Són secundàries
a
diferents patologies de l’aparell respiratori
(tuberculosis, tumors, fístules artiovenoses i
altres alteracions bronquials.
En aquests casos caldrà
diagnosticar la lesió responsable de l’hemorràgia
mitjançant broncoscòpia o exàmens
radiogràfics.
En tots els casos cal iniciar precoçment el
tractament amb el factor deficient per normalitzar la
coagulació. Cal anar a l’hospital de forma
urgent en les situacions següents:
- Hemorràgies que per la seva localització
puguin comportar importants pèrdues sanguínies
que requereixin la transfusió
de sang. En aquest
apartat s’inclouen les hemorràgies digestives,
les hemorràgies en la cavitat abdominal i els
hematomes a grans músculs.
- Hemorràgies localitzades en zones vitals: cavitat
cranial i
vies aèries.
2.3. Diagnòstic de
l’hemofília:
Es pot pensar o sospitar en el diagnòstic
d’hemofília davant de malalts amb hemorràgies
espontànies o secundàries a traumatismes,
especialment si apareixen en les primeres etapes de la
vida o en el moment del naixement. L’avaluació
detallada dels antecedents hemorràgics del malalt
i de la seva família serà un ajut important
per orientar el correcte
diagnòstic d’hemofília.
A continuació és indispensable la participació
del laboratori.
En
els malalts amb coagulopaties congènites
s’evidencien
alteracions en els proves que mesuren
globalment la coagulació.
Concretament en el cas de l’hemofília
A i B es troba allargada
la cefalina. La confirmació
del tipus d’hemofília s’obté
quan es detecta una absència o una disminució
significativa del factor
VIII (hemofília A) o
del factor IX (hemofília B).
2.4.
Diagnòstic molecular
de l'hemofília:
L'hemofília es manifesta quan en els gens
dels factors VIII o
IX
es produeixen mutacions, és
a dir, alteracions en el DNA
que donen lloc a l'absència
o pèrdua de funcionalitat de la proteïna.
Ja que els gens constitueixen el material que es transmet
de pares a fills, aquestes mutacions, i per tant la malaltia,
també
son transmeses a la descendència.
El diagnòstic molecular d'una malaltia genètica
com l’hemofília consisteix en la identificació
del defecte o mutació en el gen o
bé en
la identificació del cromosoma que és
portador del gen defectuós. Això té
una repercussió directa ja que possibilita la
realització d'un adequat consell genètic
i diagnòstic prenatal
als membres de la família
que així ho demanin.
La identificació del cromosoma portador del
gen mutat es realitza mitjançant els estudis
de lligament: s’utilitzen marcadors polimòrfics
propers als gens responsables que permeten
relacionar
un cromosoma determinat de la família estudiada
amb la manifestació de la malaltia.
Aquestes tècniques, però, tenen una sèrie
de limitacions que condicionen el seu ús, com
poden ser els casos esporàdics,
les famílies
no informatives pels polimorfismes o l'absència
d'almenys un familiar hemofílic viu.
Gràcies als recents avenços en la tecnologia
de seqüenciació
del DNA, i per tal d'evitar
les limitacions intrínseques de l'anàlisi
de lligament, s’han dissenyat protocols simplificats
per poder detectar la presència de mutacions
en els gens dels factors VIII i IX.
En el cas de l'hemofília A existeix una mutació
que és molt
més freqüent que la resta,
és l'anomenada inversió de l’intró
22. Aquesta inversió és responsable del
50% de les hemofílies A severes. Per tant, un
primer pas en el diagnòstic molecular
de l'hemofília
A severa és veure si aquesta inversió
és la responsable de la malaltia en la família.
Les tècniques emprades per detectar aquesta
mutació no requereixen la seqüenciació
i per tant poden ser realitzades
de manera més
o menys ràpida. La resta de mutacions descrites
causants d'hemofília A i B inclouen mutacions
puntuals, deleccions, insercions i estan localitzades
al llarg
dels gens.
Això obliga a determinar tota la seqüència
o composició del
gen del factor de coagulació
corresponent en el malalt i comparar-la amb una seqüència
normal per tal d'identificar
les mutacions presents.
Les mutacions trobades, com ja s’ha mencionat,
permeten el diagnòstic prenatal i consell genètic
adequats, però a més, aquestes mutacions
són enviades a bases de dades
internacionals
conjuntament amb dades clíniques, de manera
que
constitueixen una font d'estudi que permetrà
en el futur introduir millores en
les teràpies
de substitució amb factors recombinants així
com
en els assajos de teràpia gènica.
3. TRACTAMENT
DE L’HEMOFÍLIA
3.1. Concentrats de factors
de la coagulació:
Els aspectes que permeten diferenciar els diferents
concentrats
de factors de la coagulació són:
la font biològica d’obtenció (productes
derivats del plasma i productes recombinants), la puresa
i els mètodes d’inactivació viral
inclosos en el procés de producció.
Els concentrats plasmàtics s’obtenen després
de fraccionar, purificar i sotmetre el plasma a tècniques
específiques d’inactivació viral.
La qualitat i seguretat del producte final són
conseqüència de la suma de diverses etapes,
que inclouen la selecció de donants, l’anàlisi
de les donacions individuals i del plasma per fraccionament
i les etapes especifiques d’inactivació
viral.
Els concentrats de FVIII derivats del plasma es classifiquen
segons el seu grau de puresa en productes de puresa
intermèdia (PI), alta puresa (AP) i ultrapurs
(UP). La majoria d’aquests productes contenen
una quantitat rellevant d’albúmina que
s’utilitza com a estabilitzant.
Alguns dels preparats contenen factor von Willebrand
funcional, per aquest motiu són utilitzats també
en el tractament de la malaltia de von Willebrand.
Els productes de tecnologia recombinant s’obtenen
després de diverses etapes: 1. introducció
del gen que codifica el factor VIII o IX en cèl·lules
de mamífers.
2. multiplicació d’aquestes per obtenir
un banc de cèl·lules amb capacitat de
formar factor VIII o IX-
3. incubació de les cèl·lules
en un medi de cultiu apropiat perquè produeixin
el factor desitjat.
4. purificació del factor produït per
aquestes cèl·lules.
5. tractament per la inactivació viral.
6. estabilització del factor i liofilització.
Els productes recombinants tenen un elevat grau de
puresa. Depenent del procés de producció
utilitzat poden contenir
petites quantitas de proteïna
i ADN de la cèl·lula hoste, traces d’immunoglobulines
murines com a conseqüència de la purificació
del factor amb anticossos monoclonals murins i d’albúmina
humana o bovina utilitzades en el medi de cultiu.
Actualment es tendeix a la utilització de medis
de cultiu sense proteïnes d’origen humà
o animal i a la substitució de l’albúmina
humana per sacarosa com a estabilitzant del producte
final.En les taules I, II i III es recullen els diferents
factors de la coagulació actualment disponibles
en el nostre país. Taula I. Concentrats de
factor VIII
PRODUCTE Laboratori PROCEDÈNCIA PURIFICACIÓ
INACTIVACIÓVIRAL ESTABI-LITZACIÓ INDICACIÓ
HAEMATE P®Aventis Plasma humà Precipitació
Pasteurització Albúminahumana Hemofilia
A Malaltia devon Willebrand.
BERIATE P®Aventis Plasma humà Cromatografiaint.
iònic Pasteurització Sacarosa Hemofília
A.
FANHDI®Grifols Plasma humà Cromatografiaafinitat
heparina S/D +80 ºC, 72h Albúminahumana Hemofília
A.
HEMOFIL®Baxter Plasma humà Cromatografiaimmunoafinitat
S/D Albúminahumana Hemofília A.
MONOCLATE P®Aventis Plasma humà Cromatografiaimmunoafinitat
Pasteurització Albúminahumana Hemofília
A.
RECOMBINATE®Baxter Recombinant(CHO) Cromatografies
int. iònic iimmunoafinitat - Albúminahumana
Hemofília A.
REFACTO®Wyeth Recombinant(CHO) Cromatografiesint.
iònic iimmunoafinitat S/D Sacarosa Hemofília
A.
KOGENATE BAYER® BayerHELIXATE NEXGEN® Aventis
Recombinant(BHK) Cromatografies int. iònic iimmunoafinitat
S/D Sacarosa Hemofília A.
BHK= cèl·lules renals cria hàmster;
CHO= cèl·lules ovari hàmster xinès;
S/D=solvent/detergent
1 Activitat especifica excloent albúmina i FvW
2Molècula de FVIII amb delecció del domini
B
Taula II. Concentrats de
factor IX i Complex de
protrombina
PRODUCTE Laboratori PROCEDÈN-CIA PURIFICACIÓ
INACTIVACIÓVIRAL ESTABILITZACIÓ INDICACIÓ
FACTOR IX-X PBEHRINGÒAventis Plasmahumà
Adsorciósobre resina Pasteurització Estabilitzantsno
proteics Dèficit FIX, FX.
IMMUNINESTIM PLUSÒ (FIX)Baxter Plasmahumà
Cromatografiaint. iònic iint. hidrofòbica
Vapora pressió +
Detergent Estabilitzantsno proteics
Hemofília B
MONONINEÒ (FIX)Aventis Plasmahumà Cromatografiaimmunoafinitat
Tiocianat sòdic+ Ultrafiltració Estabilitzantsno
proteics Hemofília B
BENEFIXÒ (FIX)Baxter Recombinant(CHO) Cromatografiaint.
iònic Nanofiltració Estabilitzantsno proteics
Hemofília B
Taula
III. Altres concentrats de factors de la coagulació
PRODUCTE Laboratori PROCE-DÈNCIA
PURIFI-CACIÓ INACTI-VACIÓ VIRAL ESTABI-LIT-ZACIÓ
INDICACIÓ
FEIBA IMMUNO TIM 4Ò(complexe protrombina activat)Baxter
Plasmahumà Adsorciósobre resina Vapora
pressió Estabilitzantsno proteics Hemofilia A
i Bamb inhibidor.
NOVOSEVENÒNovo Nordisk Recombinant(BHK) Cromatatografiaint.
iònic iimmunoafinitat Detergent Estabilitzantsno
proteics Hemofilia A i Bamb inhibidor
3.2. Administració
del factor
En primer lloc cal seguir escrupolosament les instruccions
del fabricant del factor que estan descrites en el prospecte
del producte. Com a normes generals s’ha de tenir
en compte:
- Reservar el producte liofilitzat (l’ampolla
amb un pols blanc)
a la nevera a 4º C, no al congelador.
La resta d'elements
(aigua destil·lada, agulla
i xeringa) no cal que es posin en aquestes condicions.
- Preparar el factor en una superfície neta i
que desprès, si
cal,
es pugui netejar amb llegiu.
- Desinfectar la superfície de la pell amb una
solució desinfectant.
- Llençar les agulles i xeringues en un contenidor
rígid.
3.3. Modalitats de tractament
i dosis
El tractament substitutiu es pot fer de forma
profilàctica o només quan hi hagi hemorràgia.
En aquest últim cas, cal aplicar-lo el més
ràpidament possible. Si s’administra abans
de les primeres 4 hores, la resposta és més
efectiva i ràpida.
Per aquesta raó, s’aconsella el tractament
domiciliari. La dosi i durada del tractament dependrà
de la gravetat de l’hemorràgia. En la taula
s’exposen les guies generals per tractar les hemorràgies
més freqüents. Aquestes dades han de ser
considerades com a aproximades, ja que en cada cas cal
valorar aspectes individuals de cada malalt.
HEMORRÀGIA NIVELL
ÒPTIM FACTOR % DURADA TRACTAMENT dies
| Articular |
30 - 50 % |
1 - 3 dies |
| Hematoma muscular |
30 - 50 " |
1 - 3 " |
| Digestiva |
30 - 50 " |
2 - 3 " |
| Faringe |
40 - 60 " |
3 - 4 " |
| Retroperitoneal |
30 - 50 " |
3 - 4 " |
| Intracranial |
60 - 80 " |
7 - 10 " |
| Urinària |
30 - 50 " |
1 - 2 " |
| Menor |
20 - 30 " |
1 - 2 " |
Els concentrats de factor es poden administrar en
forma de bol (dosi única repetida segons les
necessitats) o en infusió contínua. Aquesta
última modalitat s’utilitza quan s’han
de mantenir nivells plasmàtics alts de factor
durant un període perllongat, tal com passa a
les hemorràgies agudes greus o durant les intervencions
quirúrgiques.
El tractament profilàctic s’aplica a malalts
amb deficiència
greu (factor VIII o IX inferior
a l’1%) per tal d’evitar l’aparició
d’artropatia secundària a les hemartrosis
repetides.
Es comença els primers anys de vida i consisteix
en l’administració de tres dosis setmanals
a l’hemofília A i dues
dosis a l’hemofília
B per tal de mantenir nivells mínims superiors
a l’1-2% de forma permanent.
Les dosis oscil·len entre 30 i 50 UI per kg de
pes. Moltes vegades, començar aquest tractament
tan aviat és difícil
perquè no
es disposa d’accessos venosos adequats i cal implantar-los,
la qual cosa no està exempta de complicacions,
especialment les infeccions.
3.4. Altres tractaments:
A vegades, un anàleg de la vasopresina,
l’acetat e desmopresina (DDAVP o MInurinâ),
administrat per via nasal
o intravenosa, pot ser útil
per a la profilaxis o per al tractament de les hemorràgies
lleus en malalts amb hemofília A moderada o lleu.
Aquest fàrmac provoca un augment immediat però
transitori dels nivells de
factor VIII i no té
efectes secundaris notables, llevat de rubor facial
i conjuntival transitoris.
Els antifibrinolítics (àcid tranexàmic
o Amchafibrinâ i àcid èpsilo-amino-caproic
o Caproaminâ) són una bona opció
com
a tractament coadjuvant de les hemorràgies
de mucoses,
llevat de les hematúries. S’administra
per via oral o
intravenosa i són molt ben tolerats.
Hi ha també hemostàtics per via tòpica
com la trombina liofilitzada, colàgen i d’altres,
que no han demostrat gaire
eficàcia. En canvi,
altres substàncies preparades
comercialment (Tisucol®)
són capaces de produir un coàgul
de fibrina
i són útils en aplicar-les com a hemostàtics
sobre
les ferides.
3.5. Complicacions del
tractament amb factors de la coagulació:
En l’actualitat el nivell de seguretat
pel que fa a la transmissió de virus patògens
coneguts és molt elevada com a conseqüència
de totes les mesures que s’han establert en els
darrers anys i que s’han descrit en l’apartat
anterior. En
aquest sentit cal destacar que l’objectiu
principal del desenvolupament de nous productes és
la minimització del
risc de transmissió
de malalties.
Malgrat que els darrers anys s’ha produït
una certa alarma
amb relació a la possible transmissió
de la variant de la
malaltia de Creutzfeldt-Jakob pels
factors, en base als coneixements actuals es pot afirmar
que aquest risc és molt baix o inexistent
La formació d’inhibidors , continua sent
avui en dia una de les complicacions més greus
del tractament de l’hemofília.
Malgrat
que en un principi es va creure que la utilització
de productes recombinants s’associava a una major
incidència d’inhibidors,
els estudis recents
indiquen que la seva aparició és semblant
amb els dos tipus de factor, plasmàtic i recombinant.
Altres complicacions del tractament amb factors de
la coagulació, com poden ser les alteracions
de la funció immune o les complicacions tromboembòliques,
han estat molt minimitzades amb la utilització
de productes d’elevada puresa.
3.6. Els inhibidors:
Els inhibidors del factor VIII i IX són
anticossos que apareixen en alguns malalts hemofílics
després de rebre concentrats del factor corresponent.
Aquests anticossos són capaços de neutralitzar
el factor administrat i condicionen que el tractament
sigui ineficaç.
En l’actualitat, és la complicació
més greu dels malalts hemofílics, ja que
dificulta
molt la prevenció i el tractament de les hemorràgies.
Apareixen
en aproximadament el 25% dels malalts amb
hemofília A i
en el 5% dels afectats d’hemofília
B. Generalment, es produeixen després de les
primeres administracions de factor,
i apareixen en malalts
molt joves.
Actualment se sap que el tipus de mutació del
pacient constitueix un factor de risc per a la formació
d’inhibidors
i s’ha detectat una major incidència
d’inhibidors associada a determinades mutacions
del gen del FVIII.
Cal sospitar la presencia d’un inhibidor quan
la resposta clínica al tractament no és
l’esperada. Malgrat que l’eficàcia
del tractament sigui correcte, és recomanable
fer la determinació periòdica de la presència
d’inhibidors mitjançant anàlisis
de laboratori, basats en la capacitat que té
el plasma del malalt
de neutralitzar, en el laboratori,
el factor VIII o el factor IX.
L’activitat dels inhibidors es mesura en Unitats
Bethesda
(UB). Una UB/ml és l’activitat
d’un inhibidor capaç de neutralitzar
0.5
unitats de factor VIII o factor IX.
Es consideren inhibidors de baixa resposta els que no
superen les 10 UB, i d’alta resposta els que tenen
una activitat superior a les 10 UB.
3.7. Alternatives terapèutiques en els malalts
hemofílics amb inhibidor. Anul·lació
de l’inhibidor
Quan el títol d’inhibidor és baix
es poden utilitzar dosis més altes de factor
VIII o factor IX per al tractament de les hemorràgies.
El factor VIII porcí té les mateixes qualitats
hemostàtiques
que
el factor VIII humà
i en alguns malalts no és reconegut per l’inhibidor.
Pot produir efectes secundaris, com reaccions al·lèrgiques
i trombocitopènia, tot i haver millorat amb els
nous mètodes de purificació; actualment
s’utilitza poc.
Els concentrats de complex de protrombina activada
han demostrat la seva eficàcia terapèutica,
encara que tampoc no estan exempts d’efectes secundaris
en utilitzar-los en dosis elevades (per exemple, trombosi).
S’utilitzen en dosis de 50-100 UI per kg cada
8-12 hores. Des de fa uns anys, el factor VIIa recombinant
és una altra alternativa terapèutica molt
eficaç. El seu inconvenient és la seva
curta vida mitjana en el plasma, la qual cosa requereix
administracions cada 2-3 hores per al control de les
hemorràgies.
Tal com passa en els malalts sense inhibidor, cal utilitzar
mesures complementàries com el repòs,
immobilització, aplicació de fred local,
etc.
Com que les alternatives terapèutiques en els
malalts amb inhibidor són més limitades,
un cop diagnosticada la presència
de l’inhibidor,
cal plantejar-se la seva anul·lació; és
l’anomenat tractament d’immunotolerància.
Consisteix a administrar injeccions repetides (generalment
diàries) del factor de coagulació que
manca (VIII o IX) amb la finalitat que el sistema immunitari
del propi malalt es faci tolerant al factor deficitari,
la qual cosa s’aconsegueix en un
75-85% dels malalts
tractats.
4. ALTRES
RECOMANACIONS
4.1. Higiene oral:
És evident que en els malalts amb coagulopaties
les intervencions odontològiques, incloent-hi
les més senzilles o rutinàries, com les
obturacions o empastos dentals,
representen un problema
a tenir molt en compte atès el risc hemorràgic.
Per aquest motiu, els malalts hemofílics, sovint,
es troben
amb dificultats per ser atesos a les consultes
convencionals.
Com a conseqüència d’aquests fets,
la prevenció de les principals patologies orals,
càries i malaltia periodontal (gingivitis i periodontitis),
en el malalt hemofílic adquireix
una transcendència
molt superior a la de les altres persones.
Podem assegurar que si en la població normal,
sense
patologies prèvies, aquesta prevenció
és important, en el
malalt hemofílic,
especialment si coexisteixen infeccions víriques,
passa a ser un objectiu fonamental.
Les patologies que hem esmentat anteriorment, són
altament susceptibles a les mesures preventives. Les
principals mesures preventives de la cavitat oral estan
basades en els hàbits d’higiene oral diaris.
És important destacar que no s’ha demostrat
fins al moment que l’hemofília predisposi
a patir amb més severitat càries o gingivitis
(inflamació de les genives). Normalment, en el
malalt hemofílic així com en els altres
malalts odontològics,
el sagnat de les genives
es produeix més freqüentment per
la inflamació
dels teixits gingivals, conseqüència de
la manca d’un raspallat eficaç, que per
l’alteració de la coagulació coexistent.
Com a norma general, els nens i els adults han de fer
un
mínim de dos raspallats dentals diaris, al
matí i a la nit, de
tres minuts de durada cada
un, utilitzant una pasta dental amb fluor.
L’ús de solucions especifíques
a base de fluor o altres substàncies que reforcin
la salut de les genives seran indicades únicament
pel professional a cada malalt, en particular segons
l’edat i la patologia present.
Cal aconsellar també una visita periòdica
a l’odontòleg. Semestrals o anuals, depenent
de l’edat del malalt i del risc detectat a patir
alguna de les principals malalties orals.
En aquells casos en què calgui fer un tractament
en la cavitat oral, és indispensable que existeixi
una bona comunicació
entre l’hematòleg
i l’odontòleg per establir un protocol
d’actuació que tingui en compte el risc
de la intervenció i la possible necessitat d’administrar
factor.
Des d’un punt de vista odontològic, es
consideren de risc les intervencions següents:
- la neteja dental que requereix aprofundir per sota
les genives,
- l’anestèsia troncular, utilitzada amb
freqüència en el tractament dels molars
inferiors,
- les extraccions dentals (exodòncia).
Les obturacions o empastos dentals són intervencions
de baix risc, sempre que no requereixin un grau d’anestèsia
profunda.
Malgrat que la col·locació de pròtesis
dentals i aparells d’ortodòncia no són
manipulacions de risc, cal fer el control adequat dels
aparells amb la finalitat de prevenir l’aparició
d’úlceres i lesions que comportin hematomes
o altres hemorràgies en la cavitat oral.
4.2 Exercici físic:
La pràctica d’exercici físic i esport
és important en el malalt amb hemofília
com una forma amena de mantenir el to muscular que protegeix
les articulacions. A més, és també
una excel·lent forma de relació social
amb altres nens o adults.
En primer lloc és fonamental escollir una activitat
satisfactòria per a cada malalt i que la seva
pràctica no suposi un risc superior al possible
benefici. Com a norma general, no hem de sotmetre les
articulacions amb lesions prèvies a mobilitzacions
que forcin l’arc articular o a resistències
que provoquin una sobrecàrrega.
La potenciació muscular, bàsica per a
la prevenció de la patologia articular, es pot
obtenir, sense complicacions, mitjançant la pràctica
d’exercicis isomètrics que contreuen el
múscul sense mobilitzar l’articulació
adjacent.
Naturalment és possible anar més enllà,
i en aquest sentit la Federació Mundial d’Hemofília
aconsella els següents esports:
- Natació
- Tenis taula (ping-pong)
- Senderisme
- Pesca
- Dansa
- Golf
- Bitlles
- Bàdminton
- Ciclisme
- Paddle
Per norma general, es consideren perillosos tots els
esports
que comporten una certa violència (boxa,
motociclisme) o contacte físic (handbol, bàsquet).
Malgrat que el futbol és l’esport que
més es practica, especialment a les escoles,
no és recomanable atès el risc de contacte
directe amb la pilota i el continu contacte, sovint
violent, entre els jugadors.
Abans d’iniciar una activitat esportiva, cal
fer un període d’escalfament per tal de
prevenir lesions musculars, i en finalitzar es completa
la sessió amb suaus exercicis d’estirament.
És important utilitzar un calçat adequat
que controli bé el
peu i que i l’impacte
de taló.
En cas de molèsties articulars o musculars,
per petites que siguin, cal interrompre immediatament
l’activitat. Si
existeixen lesions articulars
o musculars, és indispensable esperar la seva
complerta resolució abans d’iniciar novament
l’activitat esportiva.
Els malalts que estan en tractament profilàctic
poden aprofitar els períodes amb bons nivells
de factor per desenvolupar aquestes activitats esportives,
amb menys risc de presentar complicacions hemorràgiques.
En la pràctica d’un esport per part dels
malalts amb hemofília, cal trobar un equilibri
entre el benefici esperat, com a conseqüència
de l’activitat física i la potenciació
muscular, i
les possibles complicacions.
Caldrà seleccionar una activitat prou atractiva
per al malalt
però que s’adapti a la possible
patologia articular existent.
Atès que la majoria
dels malalts en edat de creixement segueixen pautes
de tractament profilàctic, és recomanable
coordinar el tractament i la pràctica d’esport,
de manera que
la màxima activitat coincideixi
amb els nivells més alts de factor.
5. Teràpia
gènica en l'hemofíla:
La teràpia gènica és una manera
nova de tractar les malalties basada en la introducció
de material genètic dins de les cèl·lules
per tal de corregir un problema biològic. Els
gens són fragments d'ADN que contenen la informació
per a la formació de les proteïnes necessàries
per al funcionament normal de tot l’organisme.
La teràpia gènica pretén corregir
la malaltia proporcionant a
les cèl·lules
informació perquè siguin elles mateixes
les que fabriquin el producte actiu (la proteïna),
en lloc
d’administrar-lo directament com es fa
amb els fàrmacs convencionals. En el
cas de l’hemofília
s’administraria al
malalt el gen del factor de
coagulació que li manca en lloc de donar-li el
factor.
Malgrat que en el futur serà possible reparar
els errors que existeixen en un gen i donen lloc a la
malaltia, els objectius actuals són més
modestos. Actualment, la teràpia gènica
tracta
de corregir el defecte en la funció d’un
gen anòmal introduint una còpia normal
d’aquest en les cèl·lules.
La recerca actual està centrada a desenvolupar
gens més eficaços, i vehicles o vectors
que s’encarreguen de fer que el material genètic
entri dins les cèl·lules.
Els vectors més eficaços perquè
l’entrada de material genètic sigui en
quantitat suficient són els de tipus viral. En
ells s’eliminen els gens fonamentals per la seva
multiplicació i se substitueixen pel gen terapèutic
per tal d’aprofitar la seva capacitat d’entrar
dins les cèl·lules.
Fins al moment s’han realitzat quatre assaigs
clínics en humans, en els quals s’han utilitzat
diferents estratègies.
Dels resultats obtinguts
en ells s’ha pogut deduir que les estratègies
utilitzades
i les dosis subministrades són segures.
En el primer assaig, que va tenir lloc fa 10 anys a
la Xina, es van tractar dos germans amb hemofília
B greu amb cèl·lules pròpies prèviament
extretes i a les quals s’havia introduït
el gen del factor IX.
Així es va aconseguir una persistència
de l’activitat de més
d’un any. Fins
al novembre de 1998 no es va realitzar el següent
assaig, en aquesta ocasió va ser amb hemofília
A.
L'empresa Transkaryotic Therapy, Inc va tractar els
malalts
amb cèl·lules pròpies on
s’havia introduït el gen del factor VIII,
aquest cop sense l’ajut de vectors virals.
Els resultats s’han publicat aquest any i mostren
que amb aquesta estratègia es poden aconseguir
nivells d’entre l’1-2% respecte als normals.
A mitjans de 1999 es van iniciar dos assaigs més,
en aquests casos aplicant els vectors virals directament
en els pacients.
L’empresa Chiron va treballar en l'administració
intravenosa d’un tipus de virus amb el gen del
factor FVIII, mentre que l'empresa Avigen en el seu
assaig va utilitzar la injecció intramuscular
d’un altre virus, més petit i amb el gen
del
factor FIX. En tots dos casos s’ha observat
una disminució
de la freqüència de
sagnat.
L’any 2000 s’han iniciat dos assaigs més
els resultats dels
quals encara no es diposa.
6. 10
Punts clau en l'hemofília: ;
1. Hi ha activitats que no et son
recomanables, gaudeix de
tot
allò que sí
pots fer.
2. Aprèn a administrar-te el factor seguint les
mesures higièniques necessàries, això
et donarà autonomia.
3. El tractament precoç de les hemorràgies
pot evitar complicacions més serioses; no et
quedis mai sense factor a casa.
4. Davant la presència d'hemorràgies que
poden ser vitals
(cerebrals i faringe) posa't factor
i acudeix ràpidament a l'Hospital; consulta amb
el metge davant de qualsevol situació
de dubte.
5. No prenguis aspirina ni altres antiagregants plaquetars,
consulta amb el teu metge quins fàrmacs no pots
prendre.
6. Practica de manera habitual l'exercici físic
que et pugui ser
més útil.
7. Sigues molt cuidadós amb la higiene oral i
acudeix al dentista un cop l'any.
8. És molt important realitzar estudis genètics
per conèixer l'existència d'altres portadores
a la família.
9. El consell genètic és molt aconsellable
davant de
plantejar-se qualsevol nou embaràs
a la família.
En cas de desplaçament averigua quins són
els possibles hospitals de referència.
7. Hospitals
de referència i centres de tractament de Catalunya:
Ciutat Sanitaria Vall
d’Hebron (Sanitary
city Valle de Hebron)
Unitat d’Hemofilia - (Unit of Hemophilia)
(Hospital de Traumatologia i Rehabilitació)
Passeig de la Vall d’Hebron, s/n
08035 -Barcelona - Catalonia,
tf. 0034/ 93 274
62 07 (hemophilia Emergency 24 h.)
tf.: 34-93 274 60 00
fax: 34-93 274 90 27
Organization type: Treatment Centre
Contacto:
Dr. Rafael Parra Lopez , (Coordinator
- Coordinador)
Dra. Carmen Altisent
c-electronic: rparra@vhebron.net (Coordinador - Coordinator)
Web: www.vhebron.es
________________________________________________
Hospital Clinic i Provincial
de Barcelona
Servei d’Hemoterapia i Hemostasia (Hemofilia)
C/. Villarroel, 170
08036 - Barcelona - Catalonia,
tf.: 34 93 227 54 39 (Emergency)
34 93 227 54 39
34 93 227 54 00
Organization type: Treatment Centre of Hemopilia
Contacto: Dr. J. Monteagudo
E-mail: monteaugu@clinic.ub.es
Web: www.hospitalclinic.org
________________________________________________
Hospital de la Santa Creu
i Sant Pau
Servei d’Hematologia
Av. Sant Antoni Mª Claret, 167
08025 - Barcelona - Catalonia
tf.: 0034/ 93 347 31 33
Organization type: Treatment Centre of Hemopilia
Contacto: Dr. J. Fontcuberta
c-electronic: mgari@santpau.es
Web: www.santpau.es
________________________________________________
Hospital de Sant Joan
de Déu
Servei d’Hematologia - Hemofilia - Laboratori
Ctra. d’Esplugues, s/n
08034 -Barcelona - Catalonia
tf.: +34 93 253 21 00
Fax: +34 93 203 39 59
Organization type: Treatment Centre of Hemophilia
Contacto: Dra. Mª Teresa Toll
c-electronic: info@hsjdbcn.org
www.hsjdbcn.org/index.jsp
________________________________________________
Residencia Sanitaria
Josep Trueta (Sanitary residence)
Servei d’Hematologia - Hemofilia
Avda. de França, s/n
17005 - Girona - Catalonia,
tf. 0034/ 972 20 27 00
Treatment Centre of Hemopilia
Contacto: Dra. Bustin
c-electronic: hospital@htrueta.scs.es
web: www6.gencat.net/ics/trueta/scripts/default.asp
________________________________________________ Residencia Sanitaria
Joan XXIII (Sanitary residence)
Servei d’Hematologia - Hemofilia
c/. Mallofré i Guasch, 4
43007 - Tarragona- Catalonia,
tf.: 34/977 21 77 50
tf.: 34/977 29 58 00
tf.: Admissions: 34/ 977 29 58 04
tf.: Urgències: 34/ 977 29 58 81
Treatment Centre of Hemopilia
Servei d'Hematologia i Hemoteràpia:
Contacto: Dra. Ugarriza
c-electronico: %20hospital@hj23.com
web: www.hj23.com
________________________________________________ Residencia Sanitaria
Arnau de Vilanova (Sanitary residence)
Servei d’Hematologia - Hemofilia - Laboratori
C/. Huesca, s/n
25001 - Lleida - Catalonia,
tf.: 0034/ 973 24 81 00
tf: 0034/ 973 70 22 10
Fax :0034/973 25 44 59
Treatment Centre of Hemopilia
Contacto: Dra. Araguas
c-electronico: biblioteca@arnau.scs.es
Web: www.arnau.scs.es
Resposable del Àrea d'informació mèdica:
Dr. Miquel Rutllant Bañeres, nomenat pel Patronat de la FPCH, número de col·legiat 4679, especialitat hematologia i hemoteràpia. |